Canavan Disease: Symptoms, Causes, Diagnosis and Treatment
 |
| Canavan Disease: Symptoms, Causes, Diagnosis and Treatment |
1. Definition
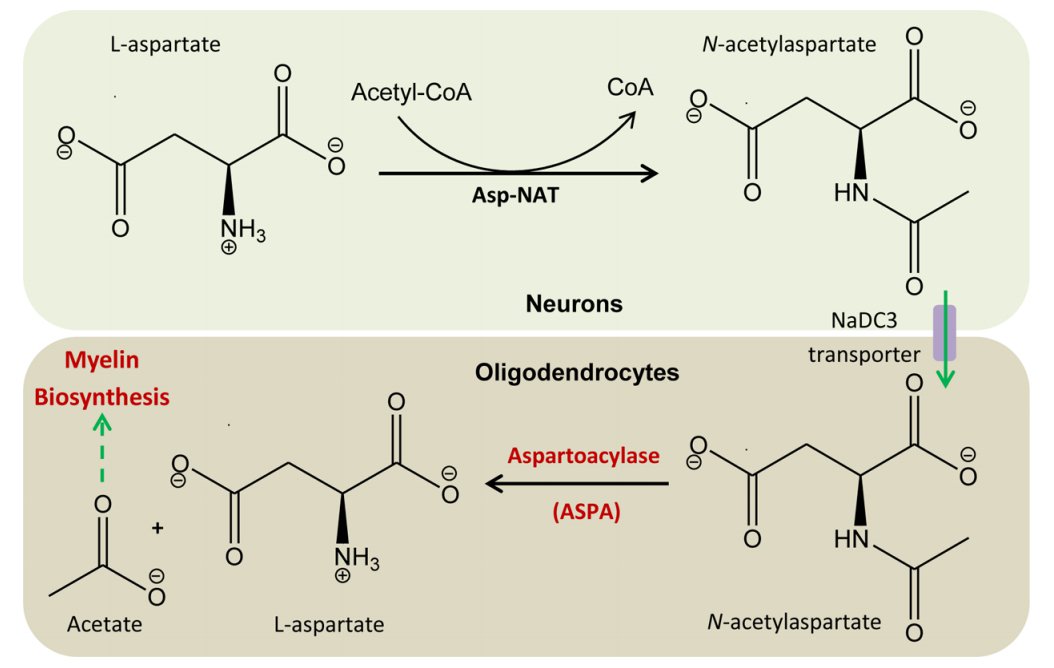
Canavan disease is a autosomal recessive neurological disorder in which the brain degenerates into spongy tissue riddled with microscopic fluid-filled spaces. Canavan disease has been classified as one of a group of genetic disorders known as the leukodystrophies. Recent research has indicated that oligodendrocytes cells in the brain responsible for making myelin sheaths, that act as insulators around nerve fibers in the brain, as well as providing nutritional support for nerve cells. For some reasons, they cannot properly complete this critical developmental task.
In Canavan disease, many oligodendrocytes do not mature and instead die, leaving nerve cell projections known as axons vulnerable and unable to properly function.
2. Etiology
Canavan disease is caused by mutation in the gene ASPA that encodes for an enzyme called aspartoacylase, which acts to break down the concentrated brain chemical known as N-acetyl-aspartate (NAA). Because of the mutation, aspartoacylase cannot be produced, leading to impairment of NAA degradation process
3. Symptoms
Symptoms of Canavan disease usually appear in the first 3 to 6 months of life and progress rapidly. Symptoms include lack of motor development, feeding difficulties, abnormal muscle tone (weakness or stiffness), and an abnormally large, poorly controlled head. Paralysis, blindness, or hearing loss may also occur. Children are characteristically quiet and apathetic. Although Canavan disease may occur in any ethnic group, it is more frequent among Ashkenazi Jews from eastern Poland, Lithuania, and western Russia, and among Saudi Arabians.
4. Diagnosis
Canavan disease can be identified by a simple prenatal blood test that screens for the missing enzyme or for mutations in the gene that controls aspartoacylase. Exceeding NAA concentration can also be identified by gas chromatography-masspectrometry. Both parents must be carriers of the defective gene in order to have an affected child. When both parents are found to carry the Canavan gene mutation, there is a one in four (25 percent) chance with each pregnancy that the child will be affected with Canavan disease.
5. Treatment
Canavan disease causes progressive brain atrophy. There is no cure, nor is there a standard course of treatment. Treatment is symptomatic and supportive.
6. Prognosis
The prognosis for Canavan disease is poor. Death usually occurs before age 10, although some children may survive into their teens and twenties.
7. What research is being done?
The gene for Canavan disease has been located. Many laboratories offer prenatal screening for this disorder to populations at risk. Scientists have developed animal models for this disease and are using the models to test potential therapeutic strategies. Three strategies are currently under investigation: 1) gene transfer using viral vector to the brain in order to replace the mutated gene for the enzyme (Leone 2012; Jonquieres 2013; Ahmed 2016); 2) metabolic therapy to provide a crucial missing metabolite (acetate) (Arun 2010; Gessler 2017); and 3) enzyme therapy where the enzyme aspartoacylase is engineered to be able to enter the brain and is injected in the the blood stream (Jonquieres 2018).
What are the symptoms of Canavan disease?The symptoms of the disease can vary greatly. Children affected by the condition may not share the same symptoms. Some of the most common symptoms are: larger-than-normal head circumference poor head and neck control reduced visual responsiveness and tracking unusual muscle tone, leading to stiffness or floppiness unusual posture, with legs often kept straight and arms flexed difficulty eating, with food sometimes flowing up into the nose difficulty sleeping seizures An increase in head circumference typically develops abruptly. Other symptoms develop more slowly. For example, vision problems may become more obvious as a baby’s development slows down. Canavan disease is a progressive condition, meaning its symptoms can worsen over time. Who is at risk of having Canavan disease?Anyone can be a carrier of Canavan disease and not have any symptoms. When both parents are carriers, each child has a 25% of having the disease. The carrier rate for the general population is 1/300. Ashkenazi Jews are at high risk with a carrier rate of 1/55. Coping with Canavan diseaseThere is no treatment or cure for Canavan disease but there are ways to manage symptoms. NTSAD is the largest organization dedicated to supporting families with members having Canavan, GM1, Sandhoff, or Tay-Sachs - allied diseases that have common symptoms and ways to manage them. We have compiled many resources for family members, provide weekly and monthly updates on events and research, and organize an annual family conference specifically for family member |
Canavan Disease Therapeutics Market by Projections, Estimations, Business Competitors Future Prospects and Forecast 2027The report presents a thorough overview of the competitive landscape of the global Canavan Disease Therapeutics Market and the detailed business profiles of the market’s notable players. Threats and weaknesses of leading companies are measured by the analysts in the report by using industry-standard tools such as Porter’s five force analysis and SWOT analysis. The Canavan Disease Therapeutics Market report covers all key parameters such as product innovation, market strategy for leading companies, Canavan Disease Therapeutics market share, revenue generation, the latest research and development and market expert perspectives. For Sample Copy of Reports: https://www.contrivedatuminsights.com/request-sample/102816 It highlights the recent market trends, growth in the past decade, and upcoming opportunities in front of the business. The research methods and tools used to analyze the studies are both primary and secondary research. The study further presents details on the funds initiated by different organizations, and industries. Source: Neighborwebsj.com |
About Author: This document was written by PhD Nguyen Hong Khanh - Researcher in Gene technology, Vinmec Research Institute of Stem Cell and Gene Technology.